With advancements in LC-MS instrumentation and methods for targeted quantification, proteomic productivity is moving from protein identifications and Venn diagrams, to establishing quantitative differences between proteins in samples representing a challenge or disease state, vs. samples representing a normal or control state. Peptides of interest are usually selected from discovery investigations and can serve as proxies for either total protein or functional amounts. For this, targeted LC-MS methods quantify peptides that meet both specific parent and daughter ion criteria, that correspond to the target peptides of interest. The terms Selected Reaction Monitoring (SRM), Multiple Reaction Monitoring (MRM) and more recently Parallel Reaction Monitoring (PRM) all fundamentally exploit this label-free data acquisition method. Furthermore, targeted LC-MS assays can be highly multiplexed, measuring up to several hundred peptides in a single acquisition. Its versatility extends to observation and measurements of functionally distinct proteoforms (i.e., active vs. inactive), post-translational modifications, along with splice and amino acid variants.
With the introduction of targeted analysis, sensitivity and technical variance in LC-MS/MS analysis has dramatically improved. Nevertheless, for serum/plasma samples, small biological variances remain hard to measure robustly. So validating small differences from discovery must translate to targeted quantification and that has become the main focus of BSG’s enrichment/depletion product line.
In blood for example, the central Complement protein, C3 circulates at ≈1500 µg/ml, while Complement Factor D circulates at ≈3 µg/ml. To measure both in one analysis requires that the MS signal intensities at both ends of the spectrum be at least reasonably proportional to the real biological concentrations, which at times can be across > 4 log ion abundance signal. Thus low abundance proteins are foremost subject to signal to noise variance, making them barely detectable and often well beyond the range where signal intensity is proportional to concentration.
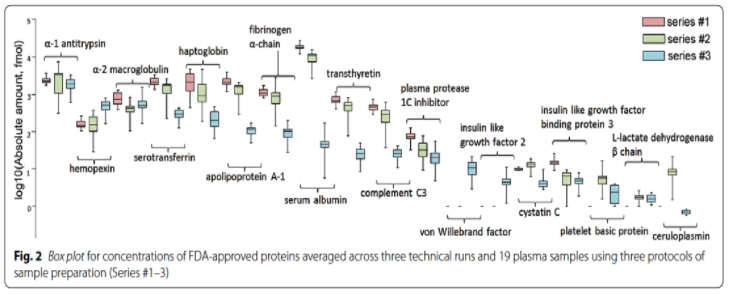
So, sample prep does matter. As an example, Kiseleva et al concluded that SRM measurements for particular proteins differed by orders of magnitude, using three protocols of sample preparation, as illustrated here.
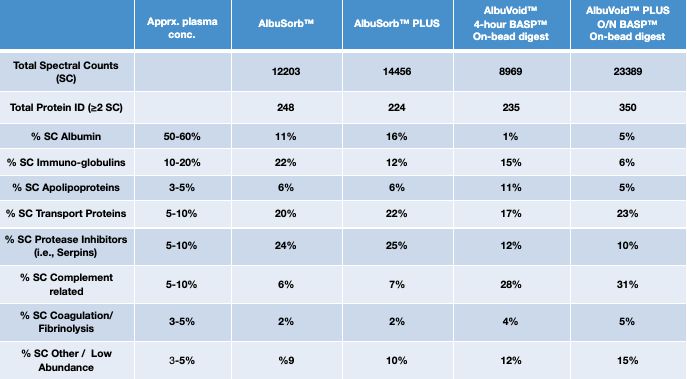
BSG’s Albumin and IgG Removal Kits offer many such options:
Digestion can be a large source of variation. A major source in serum/plasma is the high abundancy of immunoglobulins, a particularly poor digestion family of proteins, and heterogeneous in nature; it can distort digestion efficiency from sample to sample. Our recommendation is to:
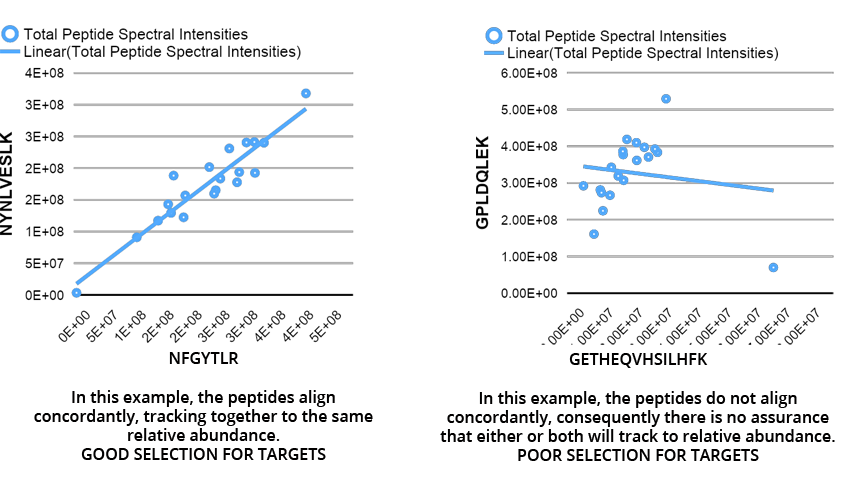
Target assays to at least two peptides from the same protein to serve as an internal control for digestion efficiency. If the peptides align to the same abundance measurement, accounting for modest technical variance, an average value provides accuracy and assurance that all sources of sample prep variance are negligible. If the peptides do not align to the same measurement, then the test may be considered suspect.
For this strategy to work, the selection of peptides becomes an important consideration. Here are two examples from our serum knowledgebase, for the same protein in different samples; in this case sp|P05546|HEP2_HUMAN, Heparin Cofactor II, Serpin D1.
As a result the selection of internal standard SIL peptides should follow several criteria: (i) they should be proteotypic - that is, the amino acid sequence should be a unique identifier to the target protein(s); (ii) efficiently ionizable in order to provide good
detection by MS; and (iii) peptides with PTMs or that might be subjected to polymorphism should be avoided, unless as in some cases, with/without may be important target information. In all cases, however, the election of the proteotypic peptides to be used as standards in quantitative proteomics is approached in an empirical way, and makes use of those peptides that have been previously observed in discovery experiments. Once selected, the LC gradients, ionization properties, and isolation widths can be varied and optimized for signal resolution and acquisition time.
CLICK TO DOWNLOAD REPORT ON HOW BSG’S SAMPLE PREP PRODUCTS HAVE BEEN CITED FOR DISCOVERY, TARGETED AND CLINICAL PROTEOMIC APPLICATIONS